Врожденная МКБ у ребенка с энтеропатией с потерей белка
Метки: Последние публикации, УЗИ почек, гастроэнтерология, генетика, дайджест, новости, педиатрия, урология
Содержание:
По данным публикации в журнале BMC Medical Genomics за ноябрь 2024: Protein-losing enteropathy with congenital kidney stones in a 2-month-old boy: a rare case report and literature review / Энтеропатия с потерей белка и врожденными камнями в почках у двухмесячного мальчика: редкое клиническое наблюдение и обзор литературы - внешняя ссылка
Энтеропатия с потерей белка (ЭПБ) — редкое заболевание, характеризующееся чрезмерной потерей белка из желудочно-кишечного тракта (ЖКТ). Сообщалось о трех основных группах расстройств, связанных с её этиологией, включая первичные эрозивно-язвенные, неэрозивно-язвенные расстройства ЖКТ и расстройства, вызывающие повышенное лимфатическое/интерстициальное давление. С точки зрения патофизиологии это состояние возникает в результате разрушения барьера слизистой оболочки кишечника или нарушения лимфатического дренажа, что приводит к чрезмерной утечке сывороточных белков в кишечник и плохой реабсорбции. Поэтому у пациентов с ЭПБ обычно наблюдается дефицит жирорастворимых витаминов, нарушение всасывания жиров, гипопротеинемия, а также недоедание.
ЭПБ проявляется в раннем младенчестве и характеризуется хронической диареей с ранним началом. Эти случаи являются частью более широкой категории врожденных диарей и энтеропатий (CODE), которые обычно связаны с пищевой непереносимостью и мальабсорбцией. Было общепризнано, что мутации DGAT1, как сообщается, участвуют в патогенезе ЭПБ и/или CODE. На самом деле, были достигнуты большие успехи в выявлении генетической основы этих расстройств с появлением и прогрессом секвенирования следующего поколения (NGS).
Камни в почках, распространенное явление у взрослых, редко встречаются у новорожденных. Насколько нам известно, после всестороннего исследования литературы не было зарегистрировано ни одного случая одновременной ЭПБ и камней в почках, о котором и сообщается в данной публикации.
10-дневный мальчик, вскармливаемый грудным молоком, поступил в больницу по причине тяжелой диареи с частотой > 10 раз в день, сопровождающейся периодической рвотой. Он родился естественным путем на 38 неделе беременности с массой тела при рождении 2,6 кг. Первоначально у пациента подозревали непереносимость кормления. При поступлении режим кормления был переведен на сухое молоко со свободными аминокислотами, на этом фоне частота диареи снизилась примерно до 2–3 раз в день, и пациент был выписан. К сожалению, частота диареи снова возросла до 5 или 6 раз в день два месяца спустя. Диарея немного облегчилась после перехода на высокогидролизованную смесь (содержание триглицеридов средней цепи 50% и без лактозы). Впоследствии у пациента появился жидкий стул, содержащий небольшое количество слизи, с частотой 4 раза в день с периодической рвотой.
Гастроинтестинальная эндоскопия выявила повышенную инфильтрацию воспалительных клеток в слизистой оболочке двенадцатиперстной кишки, с утратой щеточной каймы поверхностных ворсинок. Учитывая трудноизлечимую раннюю диарею, тяжелое недоедание и гипоальбуминемию, было настоятельно рекомендовано генетическое тестирование. После получения письменного согласия от родителей, был выполнен секвенирования всего экзома (WES) после сбора периферической крови у ребенка, периферическая кровь родителей также была собрана для секвенирования по Сэнгеру(Sanger). Результаты секвенирования по Сэнгеру подтвердили наличие гетерозиготных вариантов в гене DGAT19 (NM_012079.5) (iec276T > A и c.214 C > T), что представляло собой отцовскую унаследованную мутацию в chr8: 145 544 996 и материнскую унаследованную мутацию в chr8: 145 545 058 соответственно. На основании этого у пациента диагностирована ЭПБ, вызванная аутосомно-рецессивным вариантом гена DGAT1.
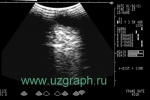
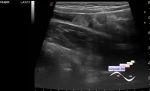
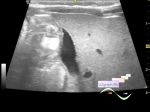
Когда пациенту было 10 дней, УЗИ почек показало множественные почечные камни, и было заподозрено, что эти камни в почках врожденные. Мальчик вновь поступил в больницу в возрасте 1 года и 4 месяцев, так как у него не было мочеиспускания в сочетании с отеком мошонки в течение 2 дней. Экстренное УЗИ и КТ-обследование показали множественные почечные камни. Позже пациент был срочно госпитализирован в ОРИТ из-за сепсиса, вызванного острой задержкой мочи и инфекцией мочевыводящих путей, инфекционной лихорадкой, электролитным дисбалансом, нарушением коагуляционной функции, камнями в почках, двусторонним гидронефрозом, дилатацией мочеточников. В ОРИТ пациент получал кислородную терапию, переливание плазмы, противоинфекционное лечение (меропенем 20 мг/кг) и противовоспалительное лечение (метилпреднизолона натрия сукцинат 1 мг/кг).
Пациенту была проведена чрескожная надлобковая цистостомия для удаления мочи перед переводом в больницу. Во время госпитализации не было рвоты кровью, крови в стуле или носового кровотечения. После начала заболевания у младенца не было рвоты при регулярном приеме высокогидролизованной смеси (содержание триглицеридов средней цепи 50%) и он плохо сосал молоко. Периферического цианоза лица и губ не было.
В последующем его масса тела составляла 5 кг, а рост — 62 см в возрасте 1 года и 7 месяцев. Сейчас у пациента по-прежнему наблюдается диарея с частотой 4–6 раз в день. Его набор веса не соответствует норме, и ему по-прежнему требуется уход за свищами. Он питается гидролизованным среднецепочечным триглицеридным молоком вместе с обычной диетой.
В этой публикации представлен случай диагностики и лечения редкого случая ЭПБ и врожденных камней в почках у новорожденного мужского пола. Из-за редкости ЭПБ диагноз ЭПБ изначально не рассматривался. У пациента изначально была диагностирована диарея младенцев, гипоальбуминемия, гипербилиарная ацидемия. Однако после длительного альтернативного использования молока со свободной аминокислотой состояние не улучшилось, и затем для дальнейшей диагностики было проведено WES. По данным авторов статьи - это первый случай мутаций DGAT1 с ЭПБ и камнями в почках.
Ген DGAT1 повсеместно экспрессируется в кишечнике человека и кодирует белок DGAT1, катализирующий конечный этап клеточного синтеза триглицеридов (ТГ). Предыдущее исследование показало, что мутации в DGAT1 могут снижать экспрессию белка DGAT1 и вызывать изменение метаболизма триацилглицеридов в фибробластах и органоидах, полученных от пациента. Хаас(Haas) и соавт. 2012г. продемонстрировали случай ЭПБ и диареи с ранним началом, содержащих мутации DGAT1. Как правило, избыточное образование диацилглицеридов или жирных кислот может быть токсичным, действуя как биоактивные сигнальные липиды или из-за детергентного поведения жирнокислотных фрагментов, что может быть тесно связано с патогенезом ЭПБ. Ван(Van) и соавт. 2018г. обнаружили, что органоиды с дефицитом DGAT1 были более восприимчивы к токсичности, вызванной липидами, что может отражать клинические особенности пациентов с ЭПБ с дефицитом DGAT1. На сегодняшний день уже описаны несколько мутаций в DGAT1. Пациенты с дефицитом DGAT1 в основном становились симптомными в течение первых нескольких месяцев жизни, что характеризовалось ранним началом, некровавой водянистой диареей, рвотой, стойкой гипоальбуминемией и задержкой развития. Недавние исследования показали, что у некоторых пациентов наблюдалась отсроченная хроническая диарея после неонатального периода, что может быть связано с их гетерозиготными мутациями в DGAT1. Для этих пациентов лечение в основном основано на ограничении энтерального потребления жира в сочетании с внутривенным введением липидов. В данном случае гетерозиготная потеря функции DGAT1 была связана с патогенезом ЭПБ, что подтверждалось фенотипами.
Мутации DGAT1 могут вызывать различные симптомы, включая нарушение всасывания липидов и дисфункцию нескольких органов. Предыдущее исследование показало, что DGAT1b может служить целевым геном NF-kB p65, который связывает сигнализацию LPS(*предположительно данное сокр. означает липополисахарид) и синтез ТГ у позвоночных. Недавние исследования показали, что накопление почечных липидов играет решающую роль в прогрессировании почечных заболеваний, включая хроническую гломерулопатию, хроническую почечную недостаточность, почечную болезнь, связанную с ожирением, а также острое повреждение почек. В отличие от предыдущих исследований, в данном случае у пациента были врожденные камни в почках в сочетании с ЭПБ. Это может быть связано с тем, что постоянная компенсация электролитов через почки может вызывать повреждение почек. У многих детей с кишечной недостаточностью наблюдалась повышенная эхогенность и нефрокальциноз, что было связано с длительным воздействием парентерального питания. Недавние исследования показали, что хроническое обезвоживание может отрицательно влиять на функцию почек. В качестве альтернативы, мальабсорбция желчных кислот, вызванная дефицитом DGAT1, может привести к диарее. Кроме того, исследования Чжао(Zhao) и соавт. 2018г. и Вана(Wang) и соавт. 2024г. выявили тесную связь между аномальным липидным обменом и образованием камней в почках. Мутации гена DGAT1 могут приводить к нарушениям липидного обмена, что может быть потенциальным механизмом образования врожденных камней в почках. У данного пациента также была гипербилирубинемия.
Действительно, ранняя диагностика и лечение имеют решающее значение в случаях диареи у новорожденных, поскольку она может быстро привести к опасному для жизни обезвоживанию и недоеданию. В данном случае WES использовался для выявления мутации в DAGT1. Генетическое тестирование определяет диагноз, лежащий в основе ЭПБ у детей, и в будущем необходимо провести больше исследований, чтобы проиллюстрировать его полезность в неонатальной диагностике.
В этой статье сообщается о редком случае ЭПБ и врожденных камней в почках у новорожденного. Дефицит DGAT1 следует с большой долей вероятности подозревать у младенцев с необъяснимой диареей и рвотой, сопровождающимися ЭПБ. WES может использоваться как эффективный метод раннего генетического обнаружения вместе с патологическим кишечным анализом.
*Также в публикации присутствуют эхограммы, данные генетического анализа, рентгенограмма, снимок КТ и микропрепарата.
*комментарии редактора